 欢迎来到乐鱼网页版登录入口网站!欢迎来到乐鱼网页版登录入口网站!
欢迎来到乐鱼网页版登录入口网站!欢迎来到乐鱼网页版登录入口网站!
15221734409


各位读者好,今天为大家带来一篇使用综合运用原代造血干细胞、人源化小鼠以及斑马鱼模型并结合前沿分子生物学技术、RNA-Seq、分子对接等研究手段来研究TMEM187调节红细胞生成以及潜在机制的高分文章,是由南京大学医学院团队2026年3月在Blood发表的,题为“TMEM187 is a novel modulator in the regulation of erythropoiesis”。深入探究了TMEM187通过与RAB11相互作用,抑制RAB11-GRAB结合,从而调控转铁蛋白受体1(TfR1)的内体循环和铁摄取效率的核心机制,为红细胞相关疾病的诊断和治疗提供理论依据。

发表杂志:
《Blood》是美国血液学会(American Society of Hematology, ASH)的官方旗舰期刊,创刊于1946年,是血液学领域被引用量最高的同行评议学术出版物,也是全球血液学领域的顶级权威刊物。

2026 年影响因子:23.9
ISSN:0006-4971
中科院分区:大类医学1区,小类血液学1区TOP期刊
发文量:每年出版文章数平均约501篇
发表成本:订阅模式无版面费,开放获取(OA)模式费用为3800–4800美元
审稿周期:首次同行评议决策平均约17天,从投稿到正式接收整体周期约2.5–4个月,接收后1–2周即可在线优先发表
《Blood》是血液学领域标杆级旗舰顶刊,学术影响力稳居全球第一梯队,是美国血液学会(ASH)官方会刊、稳定位列医学 1 区 Top SCI 期刊,在国家自然科学基金申报、国际学术评价与项目结题中认可度极高。收稿覆盖造血发育、红细胞代谢、血液肿瘤、血栓止血、免疫血液学等核心方向,尤其欢迎兼具机制原创性与临床转化价值的研究,精准匹配血液学领域全链条成果发表需求。近年国内学者发文占比稳步提升,审稿流程规范透明、评判标准公允,无隐性投稿歧视,适合深耕血液学基础与临床研究、追求高质量学术发表、希望研究成果获得全球广泛关注的科研团队投稿。
研究背景:
红细胞生成依赖于转铁蛋白及其受体TfR1介导的铁摄取,而TfR1的循环机制与细胞铁需求的关联尚不明确。TMEM187是功能未知的高尔基体跨膜蛋白,前期研究发现其可能与系统性红斑狼疮相关,且突变细胞呈现红系分化表型。鉴于高尔基体蛋白在TfR1循环中的潜在作用,本文旨在探究TMEM187是否通过调控TfR1循环参与红细胞生成,以填补铁代谢与红细胞生成调控机制的空白。
本文鉴定人类TMEM187为红细胞生成的新型负调控因子。通过细胞模型、斑马鱼和人源化小鼠实验,发现TMEM187缺失可加速红细胞成熟、铁摄取及细胞衰老,而其过表达则导致贫血。机制上,TMEM187通过与RAB11相互作用,抑制RAB11-GRAB结合,从而调控转铁蛋白受体1(TfR1)的内体循环和铁摄取效率,揭示了TMEM187在红细胞分化、成熟和衰老中的关键作用。
研究框架:
1.提出问题:
基于TMEM187突变细胞的红系分化表型,提出其是否调控红细胞生成及TfR1循环的科学问题。
2. 研究框架:
从细胞模型(K562、CD34+细胞)、模式生物(斑马鱼、人源化小鼠)层面,结合分子机制探讨TMEM187的功能。
3. 研究方法:
采用基因敲除/敲低、流式细胞术、免疫荧光、Co-IP、RNA测序、铁代谢检测等实验技术。
4. 分析数据:
通过表型观察(细胞成熟、铁摄取、贫血等)和分子互作(TMEM187-RAB11-GRAB)验证假设。
5. 研究结论:
综合细胞和动物实验结果,阐明TMEM187通过调控TfR1循环负向调节红细胞生成的机制。

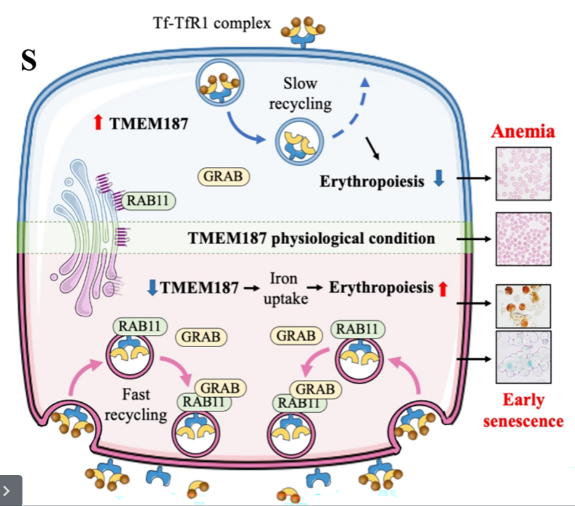
Figure 7S.机制示意图
结果解析:
1. TMEM187在体外抑制红细胞分化
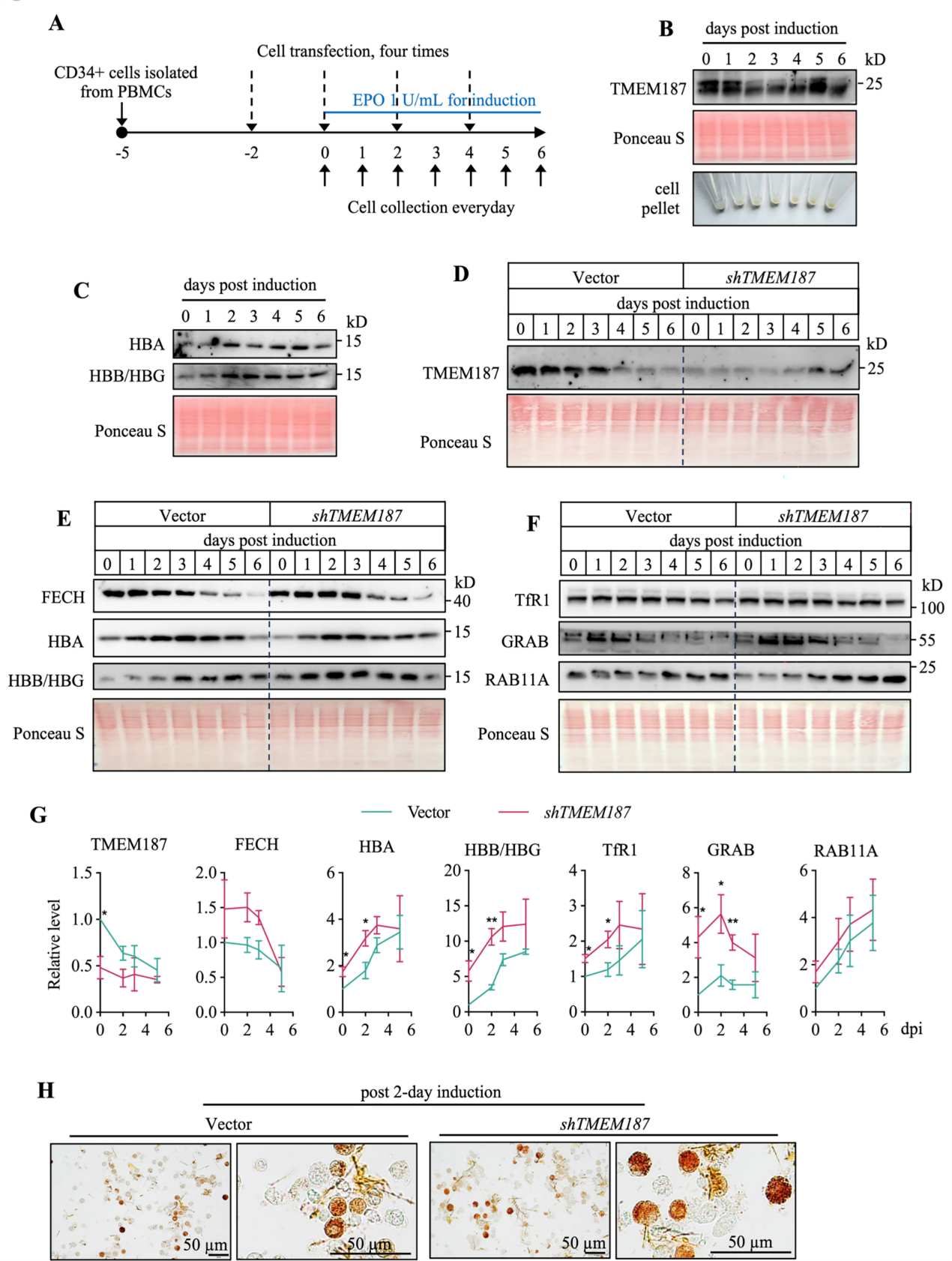
通过K562细胞模型发现TMEM187在红细胞分化过程中表达动态变化(早期降低后恢复),其敲低或敲除可促进血红蛋白合成、加速红细胞成熟,表明TMEM187是红细胞分化的负调控因子。
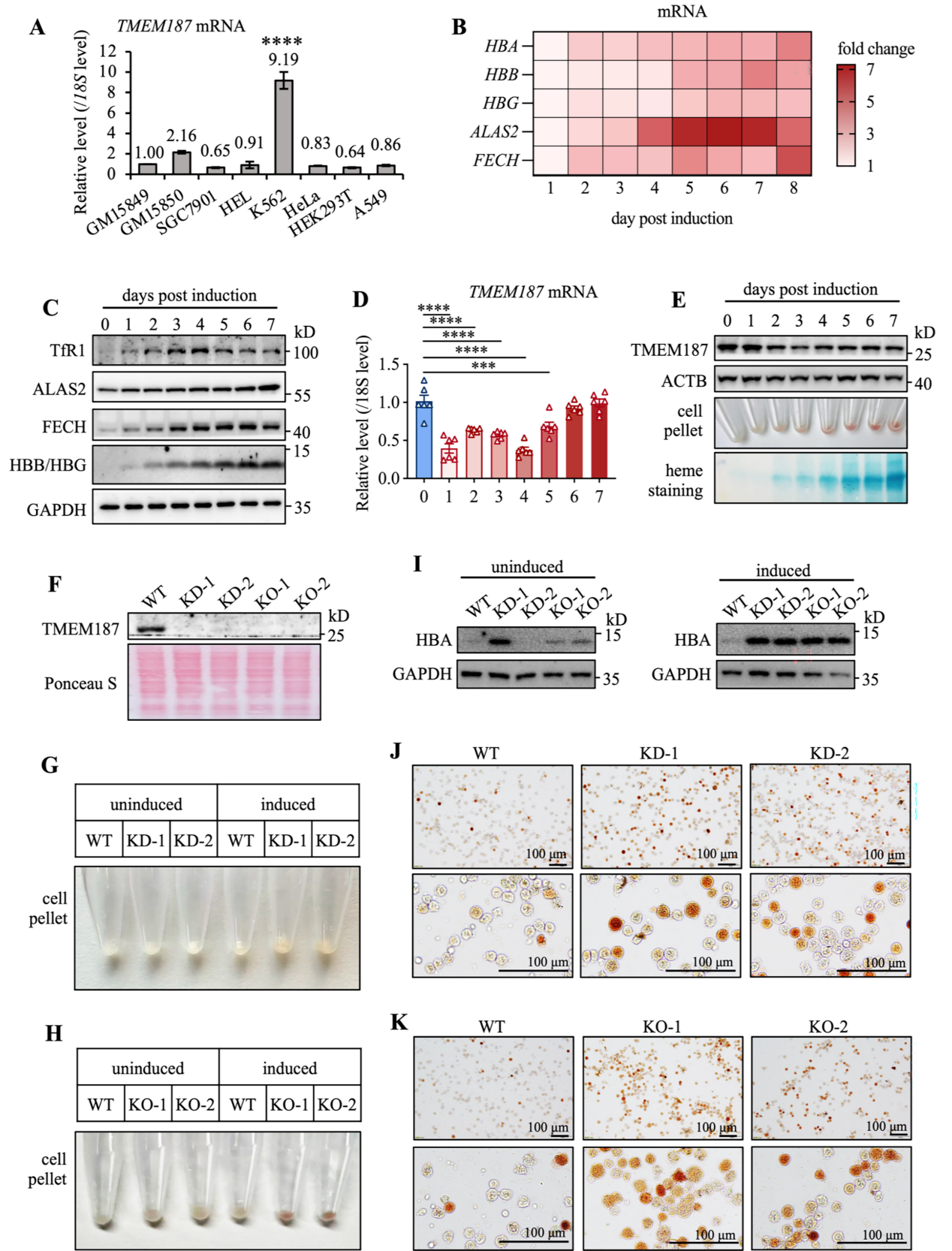
2. TMEM187缺失重编程K562细胞以促进红细胞生成
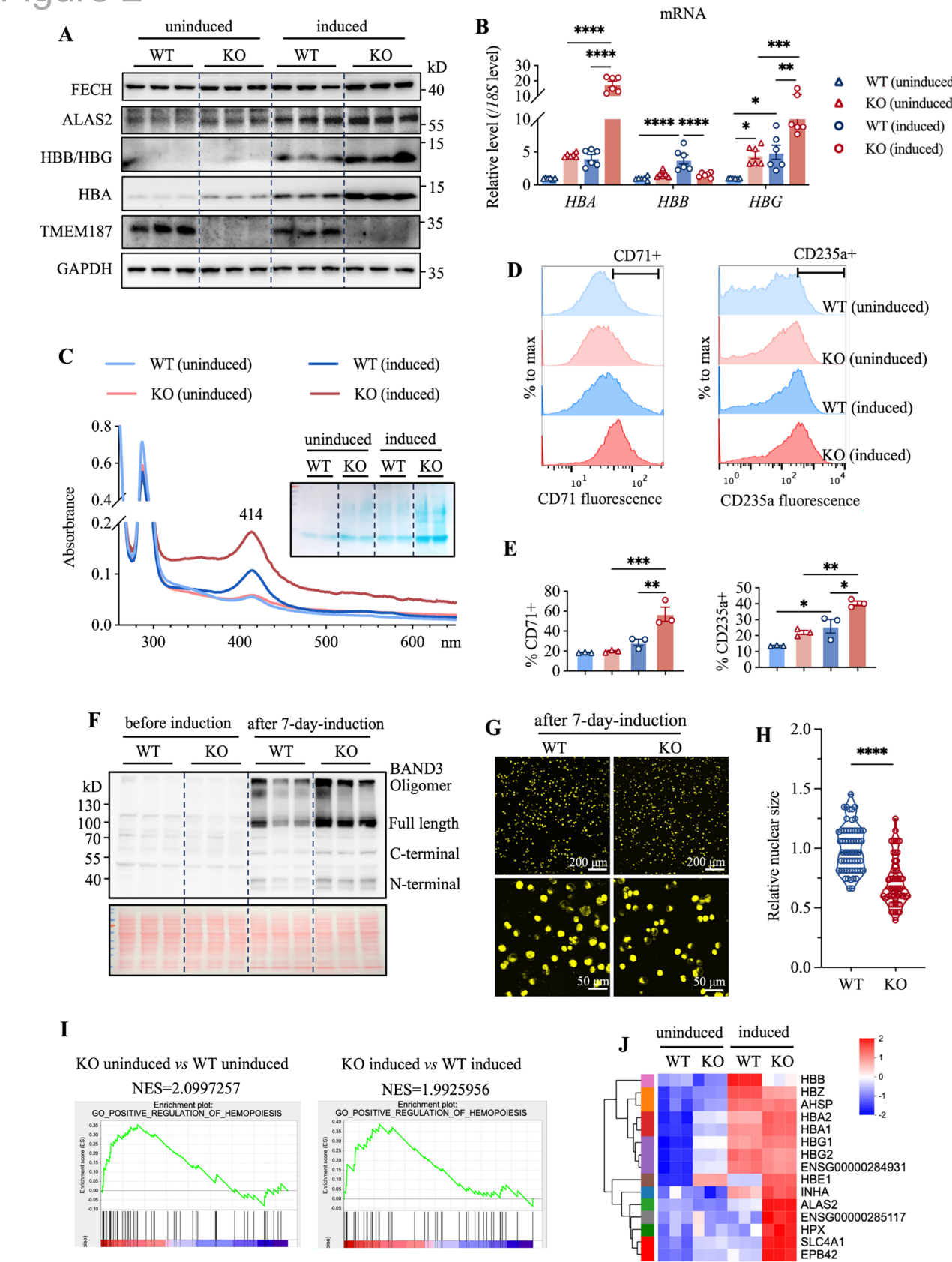
TMEM187缺失导致血红素生物合成相关基因(ALAS2、FECH)和血红蛋白亚基(HBA、HBG)表达上调,细胞表面CD71和CD235a水平升高,核体积缩小,转录组分析显示造血通路显著富集,证实其通过调控红细胞分化关键基因促进成熟。
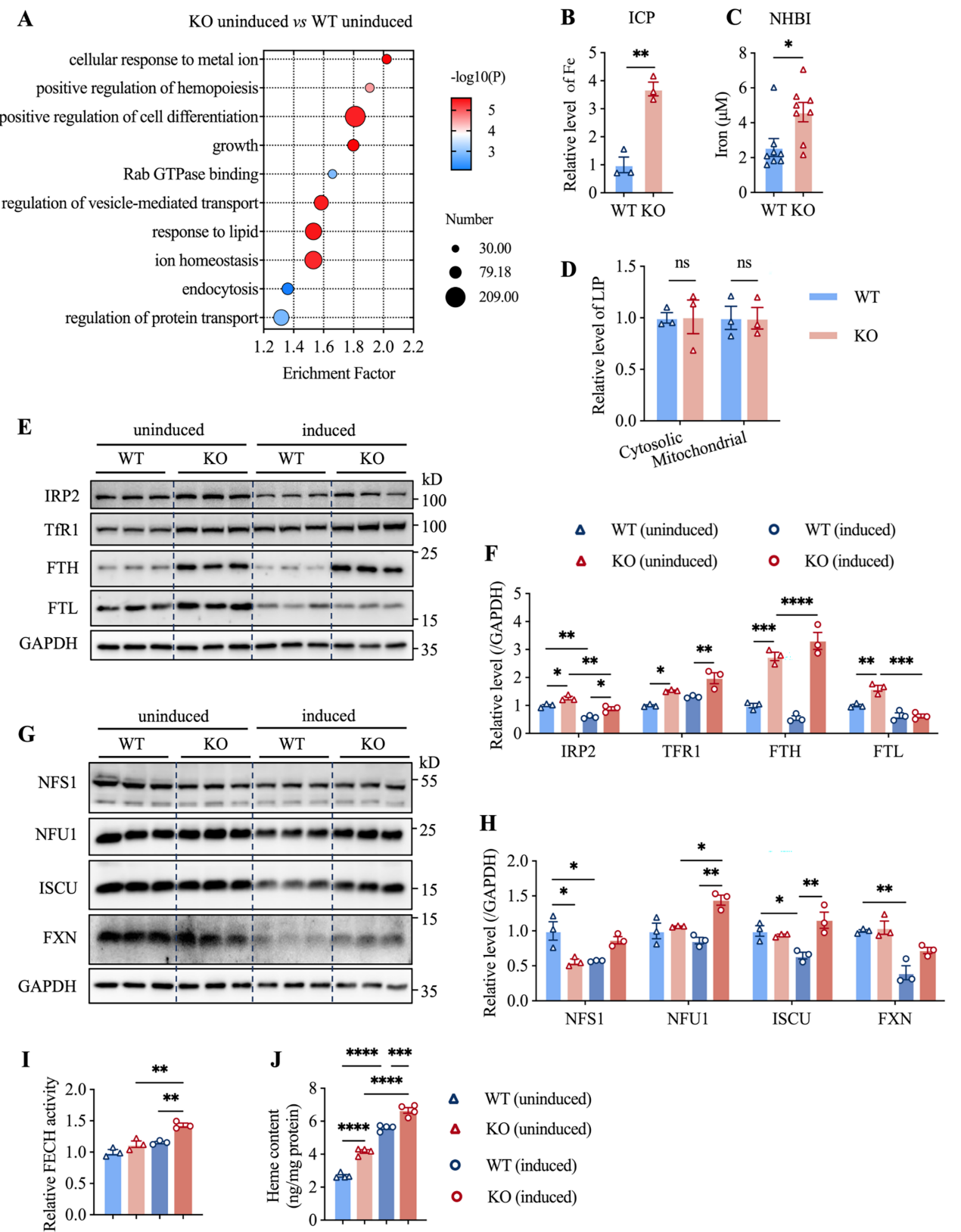
3. TMEM187缺失通过铁代谢重编程促进K562细胞红细胞分化
TMEM187缺失增加细胞总铁和非血红素铁含量,上调TfR1和铁蛋白表达,促进Fe-S簇生物合成及FECH活性,最终加速血红素合成,表明其通过调控铁吸收和利用影响红细胞生成。
4. TMEM187通过RAB11-GRAB-TfR1通路调控铁摄取效率
TMEM187与RAB11A结合(优先结合GDP结合型),竞争性抑制GRAB与RAB11A的相互作用,从而抑制TfR1循环至细胞膜。缺失TMEM187后,GRAB-RAB11A相互作用增强,TfR1循环加速,铁摄取效率提高。
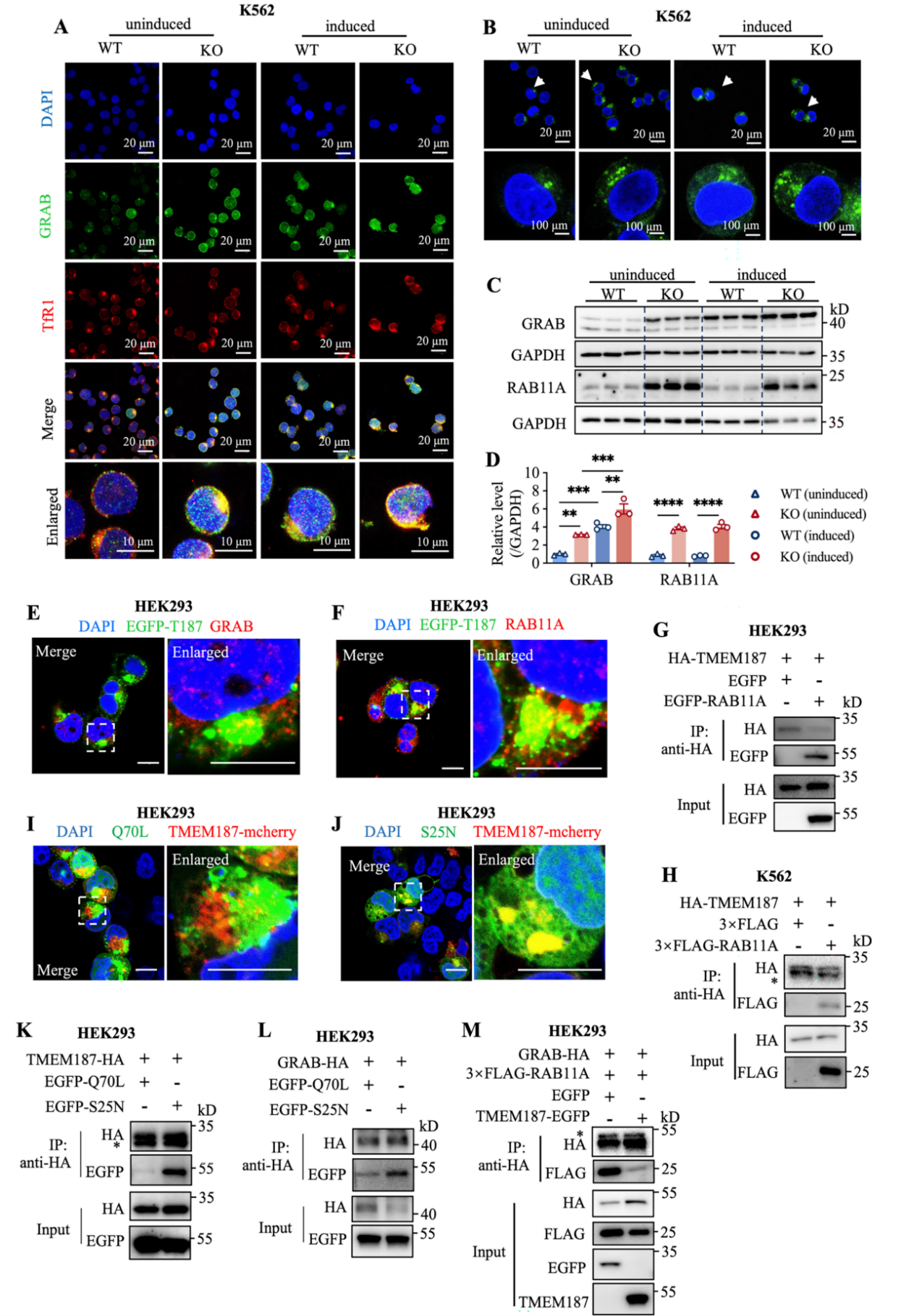
5.干扰人原代CD34+造血干细胞中TMEM187表达增强红细胞分化
在人原代CD34+细胞中敲低TMEM187,显著上调血红素合成酶(FECH)和血红蛋白(HBA、HBB)表达,促进血红蛋白化,验证了TMEM187在生理条件下对红细胞分化的负调控作用。
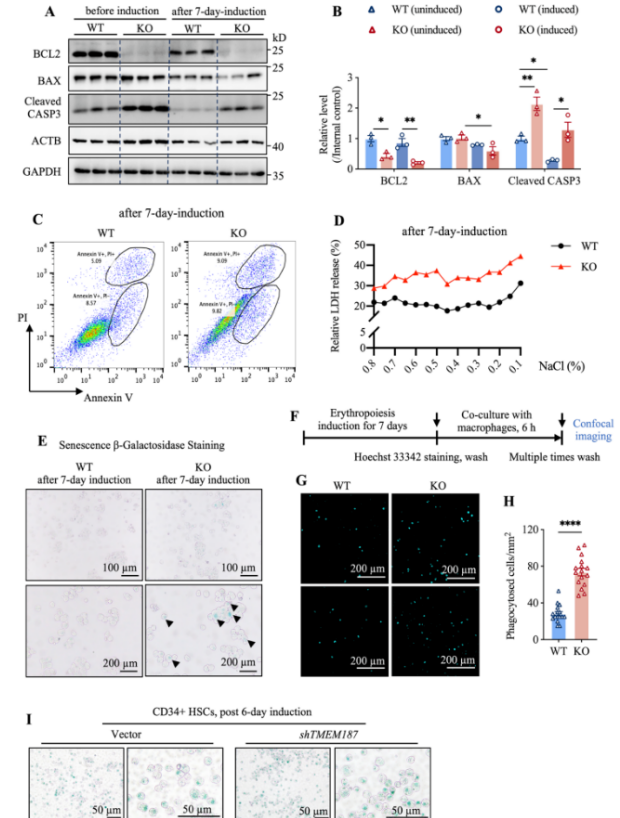
6. TMEM187缺失加速细胞衰老并增强巨噬细胞吞噬
TMEM187缺失导致红细胞提前成熟,伴随磷脂酰丝氨酸外翻、细胞膜脆性增加及β-半乳糖苷酶活性升高(衰老标志物),最终被巨噬细胞吞噬清除的比例显著增加,表明其缺失缩短红细胞寿命。
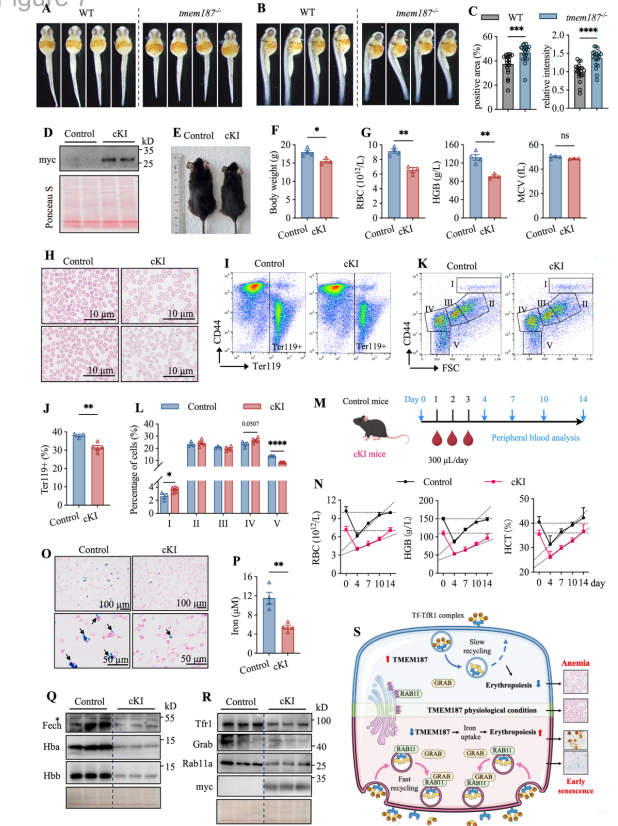
7. TMEM187在斑马鱼和小鼠模型中负调控红细胞生成的体内验证
斑马鱼tmem187敲除胚胎早期红细胞生成增强(48 hpf血红蛋白水平升高);造血谱系表达人TMEM187的小鼠出现贫血,骨髓红细胞成熟受阻,铁含量降低,应激条件下红细胞恢复延迟,证实TMEM187在体内调控红细胞生成的保守作用。
研究结论:
TMEM187是红细胞生成的新型负调控因子,通过与RAB11竞争性结合抑制GRAB-RAB11相互作用,调控TfR1内体循环和铁摄取,进而影响红细胞分化、成熟及衰老。
研究的创新性:
首次发现TMEM187的生理功能,揭示其通过RAB11-TfR1轴调控红细胞生成的新机制,为铁代谢与红细胞生成的关联提供新视角。
研究的不足之处:
未明确TMEM187在其他组织中的作用;斑马鱼成年后表型代偿机制未深入探讨;TMEM187与RAB11结合的结构基础缺乏解析。
研究展望:
后续可研究TMEM187在其他铁代谢相关疾病(如缺铁性贫血、 MDS)中的作用;探索TMEM187-RAB11相互作用的结构机制及小分子调控;利用TMEM187敲除模型研究红细胞衰老与铁再循环的关联。
研究意义:
揭示了TMEM187在红细胞生成中的关键作用,为理解铁代谢调控网络提供新靶点,可能为红细胞相关疾病的诊断和治疗提供理论依据。

