 欢迎来到乐鱼网页版登录入口网站!欢迎来到乐鱼网页版登录入口网站!
欢迎来到乐鱼网页版登录入口网站!欢迎来到乐鱼网页版登录入口网站!
15221734409


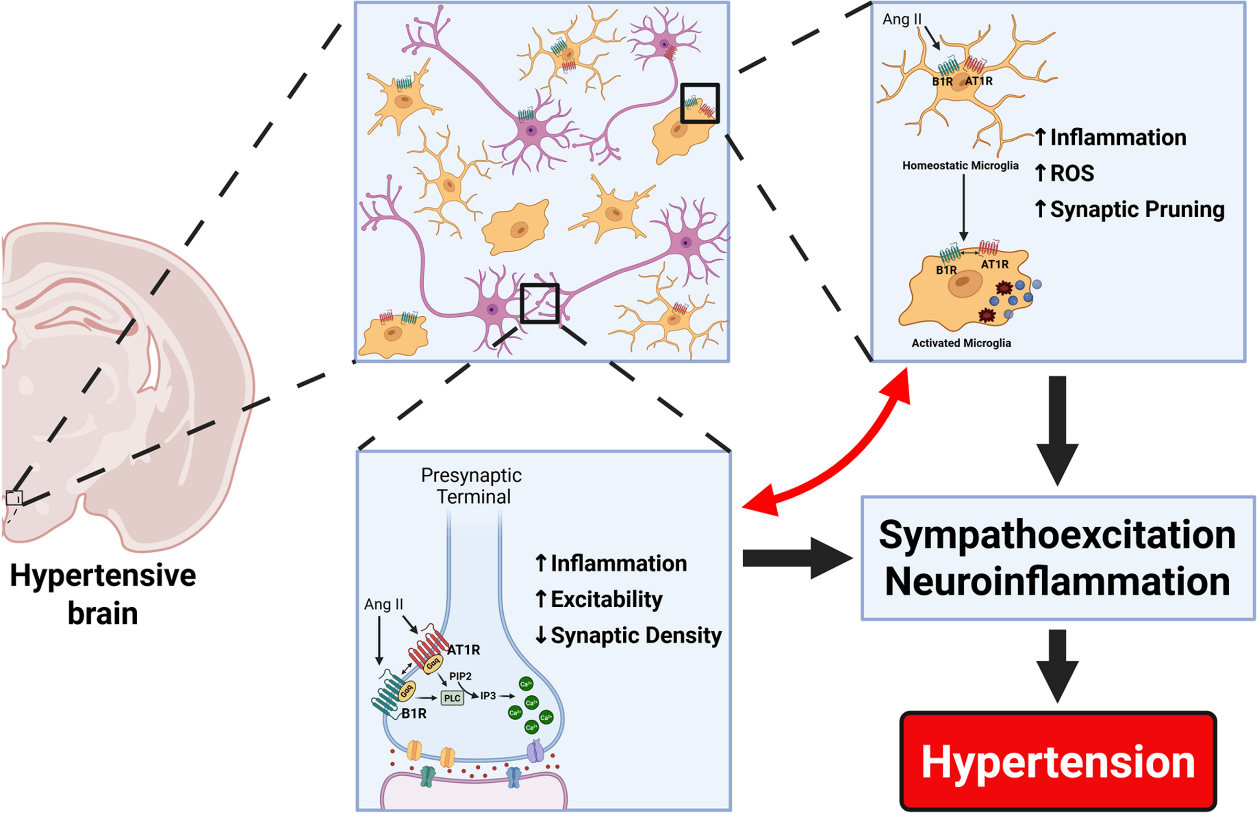
2025年8月21日,《Circulation Research》在线发表东卡罗来纳大学Sriramula团队的突破性成果。研究揭示,缓激肽B1受体(B1R)在中枢神经系统高血压“神经炎症爆发窗口”中扮演交感驱动主控角色:于Ang II输注早期,B1R在下丘脑室旁核被选择性上调,与AT1R直接组装成功能复合体,驱动神经元超兴奋、小胶质细胞激活及突触前膜丢失,从而成为神经源性高血压级联的核心引擎。

背景:
神经源性高血压以交感神经过度兴奋为核心,中枢Ang II-AT1R通路被视为始动因素,但仍有部分患者对现有RAS抑制剂反应不佳,提示存在其他放大机制。缓激肽B1受体(B1R)在炎症或损伤后迅速上调,可促炎、促交感,且前期研究发现其参与DOCA-盐高血压,但B1R是否及如何与中枢AT1R协同驱动Ang II型高血压仍未知。本文即在这一背景下,系统探讨B1R在高血压中枢的作用及其与AT1R的相互作用。
结论1: 高血压患者PVN中激肽B1R表达上调
研究发现,高血压患者脑内B1R在PVN和SFO显著上调,主要定位于神经元,并与血压水平正相关。B1R与AT1R相互作用增强,提示其在中枢血压调控中具有重要作用。
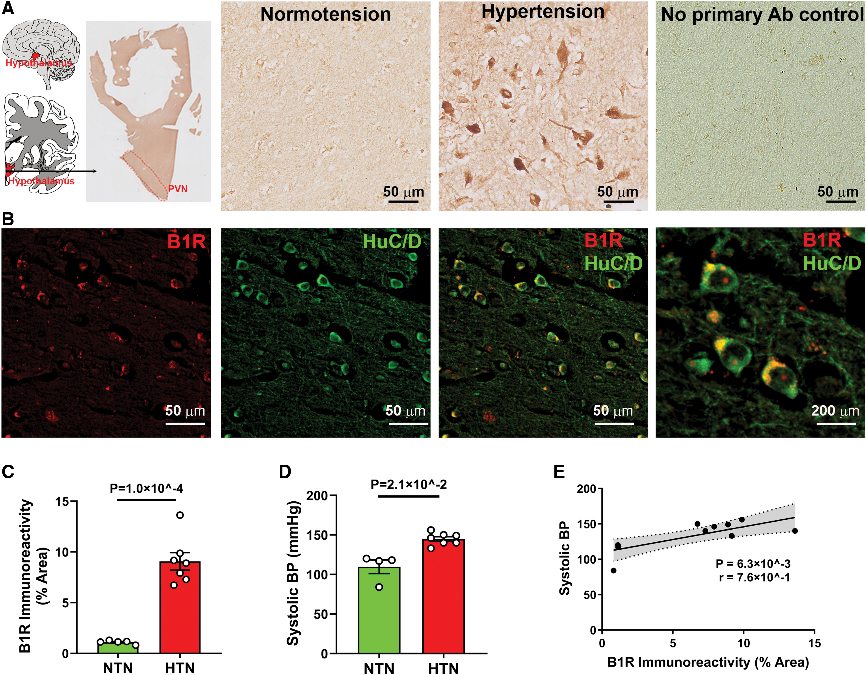
Fig1. 高血压患者脑室旁核(PVN)神经元中B1R(激肽B1受体)
免疫反应性增加
结论2: 激肽B1受体在调控血压的关键脑区中上调
Ang II诱导小鼠高血压时,激肽-缓激肽系统被激活,B1R在下丘脑血压调控核团显著上调,尤以PVN突出。B1R主要定位于神经元和小胶质细胞。脑室注射B1R激动剂DABK可引起收缩压升高,并在神经元中诱导B1R急性上调,证实B1R的中枢激活在高血压病理生理中具有关键作用。
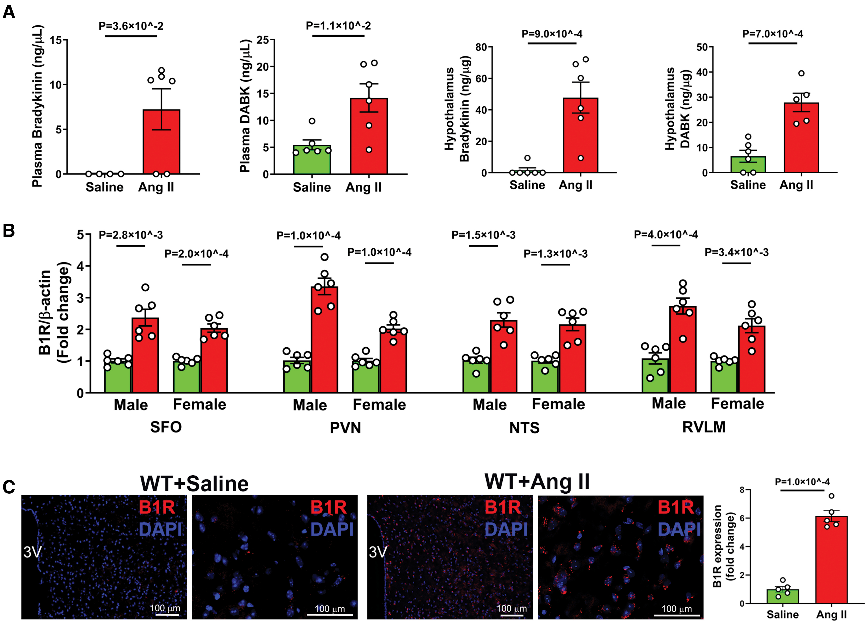
Fig2. 关键心血管调节中心中 B1R(激肽 B1 受体)表达增加
结论3:激肽B1受体缺失减轻神经源性高血压及自主神经功能障碍
研究表明,B1R缺失可显著减轻Ang II诱导的小鼠高血压,维持压力感受器反射功能,并抑制交感神经过度激活和体液调控异常。结果提示B1R在神经源性高血压的发生发展及自主神经功能紊乱中发挥重要作用。
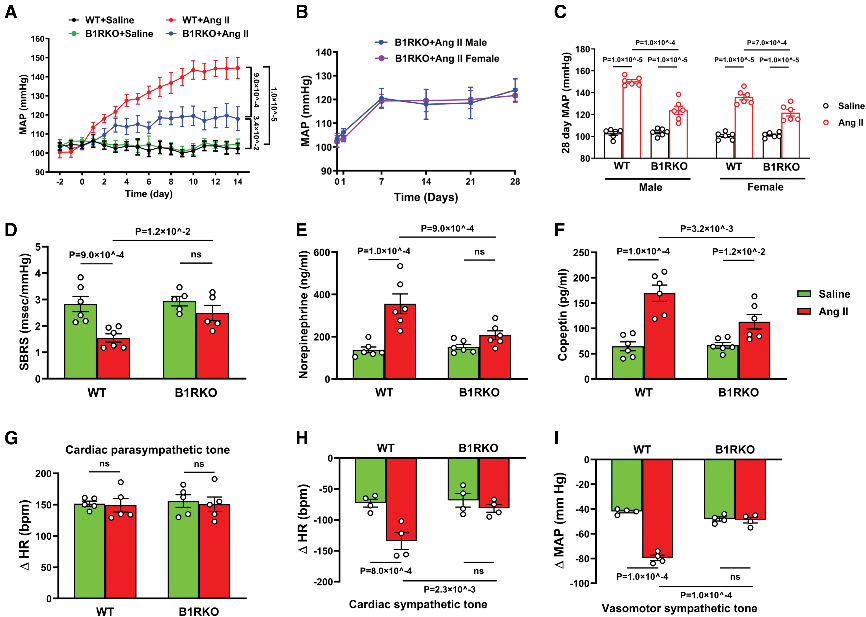
Fig3. 激肽B1受体(B1R)基因缺失可预防神经源性高血压的发展
结论4: 阻断B1受体减轻Ang II诱导的小胶质细胞活化及神经炎症
研究表明,B1R在Ang II诱导的高血压中促进小胶质细胞活化,导致促炎因子上调、抗炎信号下降,形成持续的神经炎症微环境。该过程增强神经元活性和交感驱动,加重高血压;而B1R缺失可显著减轻炎症反应和自主神经异常。
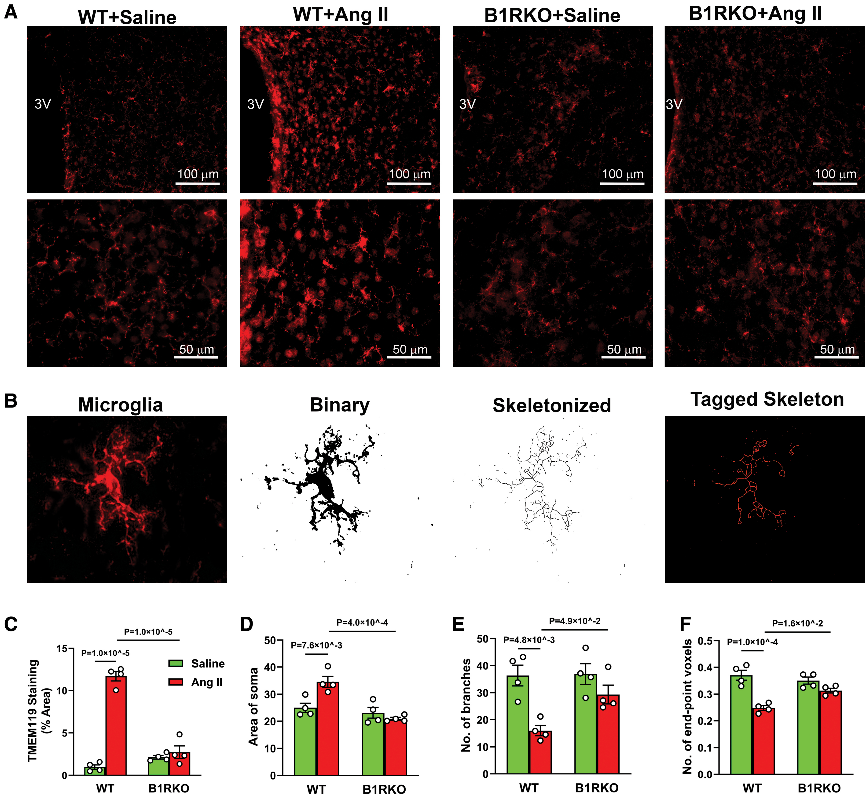
Fig4. B1R(激肽B1受体)阻断可防止Ang II(血管紧张素II)诱导的
高血压中的小胶质细胞激活
结论5:B1R的上调可诱导神经元过度活跃
B1R激活可直接增强皮层神经元放电频率和爆发持续时间,模拟高血压中交感驱动增强的特征;拮抗剂R715可显著逆转此效应。B1R对神经元同步性无影响,提示其通过增强单个神经元兴奋性而非网络整体协同来促进自主神经功能紊乱。
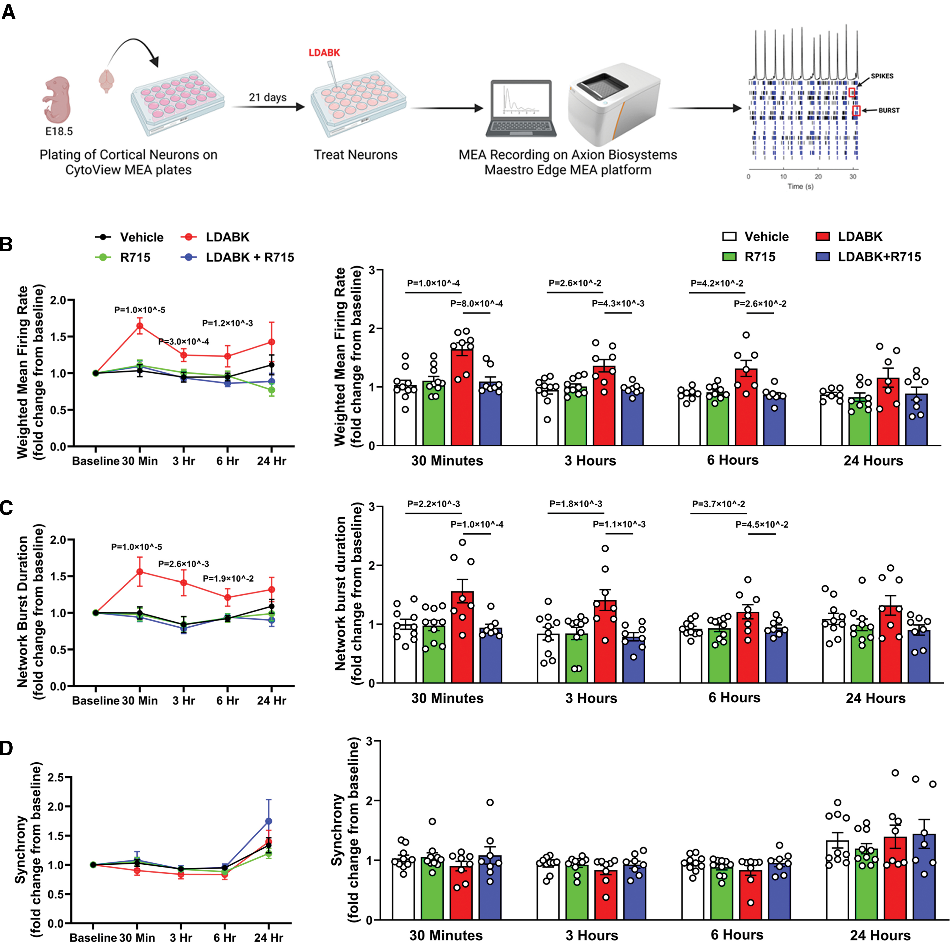
Fig5. B1R(激肽 B1 受体)刺激诱导神经元放电和爆发持续时间
结论6:B1受体定位于PVN突触前位点并促使高血压相关突触丢失
研究表明,B1R在Ang II诱导高血压中主要定位于突触前终末,促进突触前密度增加并导致突触连接失衡,而非通过神经元凋亡介导。B1R缺失可减轻突触结构异常,提示其在调控神经元兴奋性、突触传递及自主神经功能障碍中发挥关键作用。
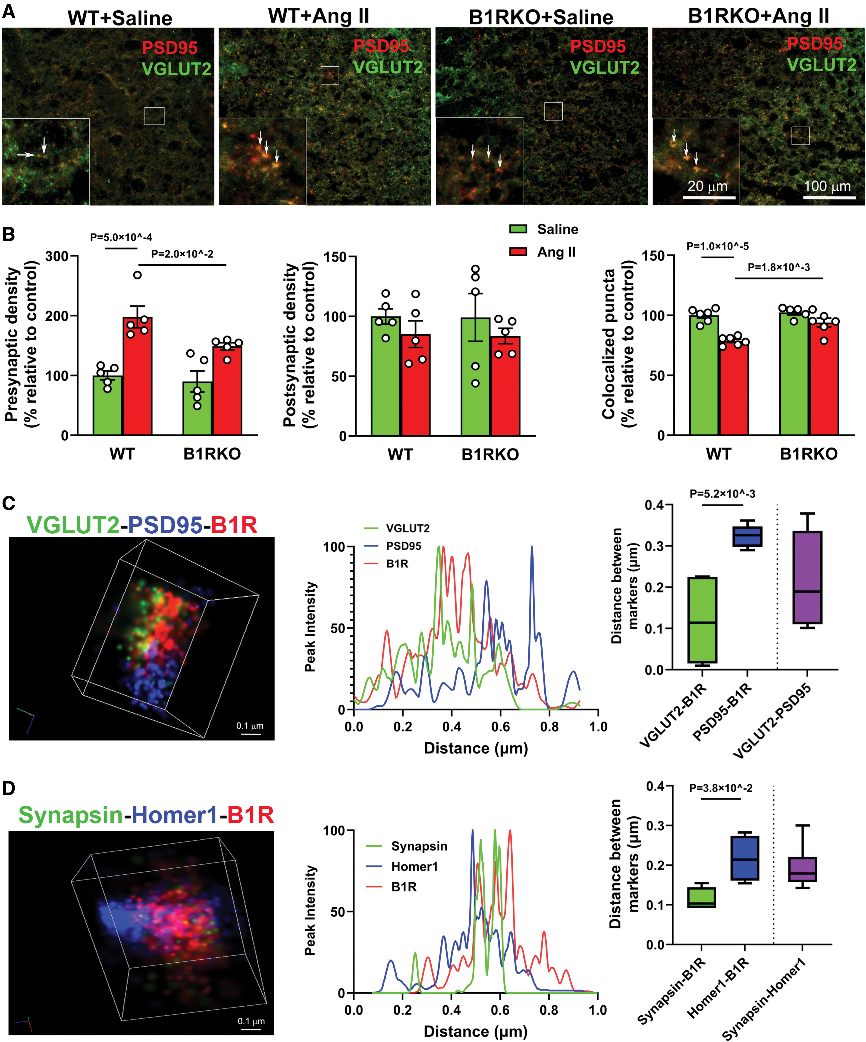
Fig6. B1R(激肽 B1 受体)阻断可减少 Ang II(血管紧张素 II)
诱导的突触丢失
结论7:高血压状态下B1受体与AT1受体相互作用增强
研究表明,B1R与AT1R在PVN形成直接相互作用,Ang II可增强此受体复合物,促进神经源性高血压。中枢B1R阻断可减轻血压升高和氧化应激,而外周阻断无效。分子对接证实B1R-AT1R通过氢键和疏水相互作用形成复合物,DABK可结合其活性位点,揭示B1R-AT1R相互作用在高血压中的分子机制。
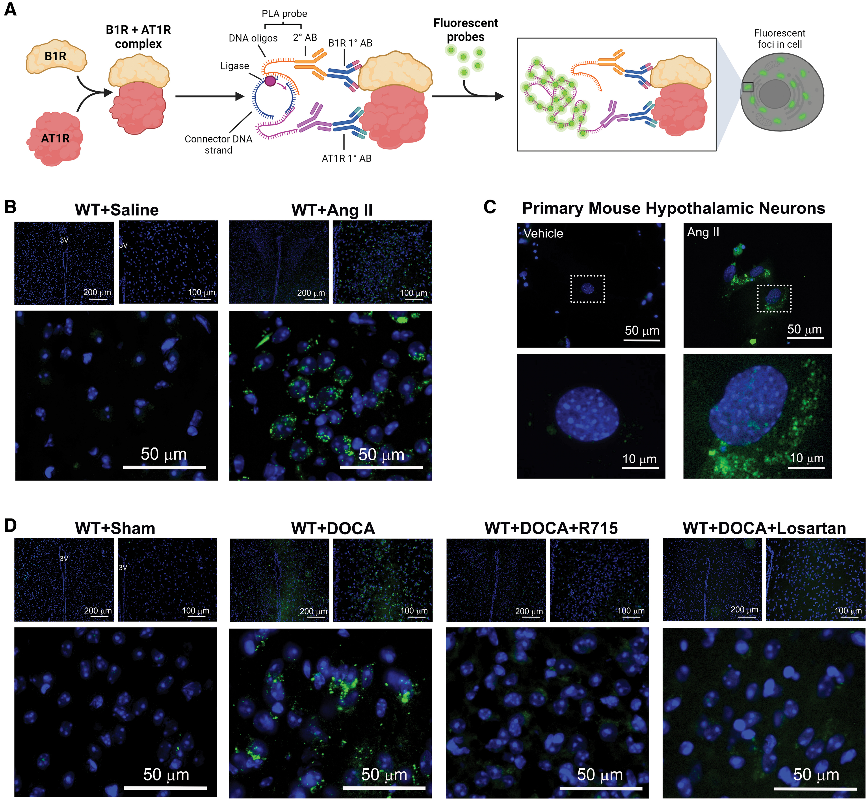
Fig7. AT1R-B1R(Ang II [血管紧张素 II] 1 型受体和激肽 B1 受体)之间的
相互作用在高血压中上调
结论8:B1受体拮抗可减轻Ang II诱导的高血压
SSR240612中枢阻断B1R可有效降低Ang II诱导的血压升高、减轻氧化应激并抑制PVN中B1R-AT1R相互作用。机制研究显示,AT1R激活可上调B1R,B1R活化反向增强AT1R表达,形成双向调控环路。这表明B1R在神经源性高血压中通过调控受体互作和信号放大发挥核心作用。
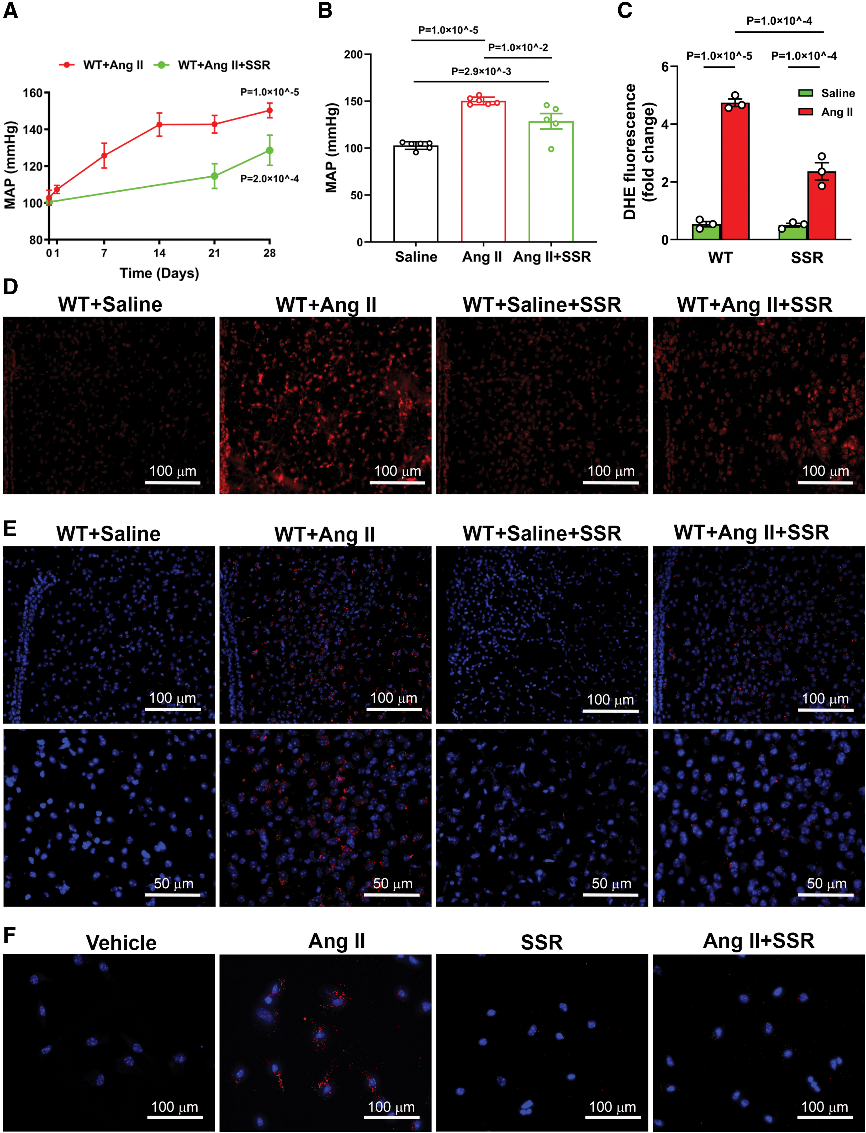
Fig8. B1R(激肽 B1 受体)的药物阻断可预防 Ang II(血管紧张素 II)诱导的高血压
总结
本研究在人高血压脑中发现下丘脑室旁核B1R表达显著升高并与血压正相关;在小鼠体内证实B1R与AT1R直接结合,形成“Ang II→AT1R→B1R→放大交感输出”的正反馈环路。基因敲除或药理学阻断B1R可显著降低Ang II诱导的高血压、恢复压力反射、抑制小胶质细胞激活及神经炎症,并防止突触前膜丢失。超分辨定位显示B1R主要位于谷氨酸能神经元突触前膜,MEA记录证实其可迅速增加神经元放电。研究提出在现有ARB基础上加用中枢B1R拮抗剂的新策略,但尚未验证高血压形成后的干预效果,且人体样本量小,机制深度与安全性仍需进一步研究。

